Aetiology, Clinical Profile and Management Dilemmas in Transgender Children with Y Containing Chromosomes: The Tip of an Iceberg
<p class="MsoListParagraphCxSpFirst" style="margin: 0in; margin-bottom: .0001pt; mso-add-space: auto; text-align: justify; text-justify: inter-ideograph; text-indent: 0in; line-height: 150%; mso-list: l0 level2 lfo1;"><!-- [if !supportLists]--><strong><span style="font-size: 10.0pt; line-height: 150%; font-family: 'Cambria','serif'; mso-ascii-theme-font: major-latin; mso-fareast-font-family: Cambria; mso-fareast-theme-font: major-latin; mso-hansi-theme-font: major-latin; mso-bidi-font-family: Cambria; mso-bidi-theme-font: major-latin;">1.1.<span style="font-variant-numeric: normal; font-weight: normal; font-stretch: normal; font-size: 7pt; line-height: normal; font-family: 'Times New Roman';">Â Â Â Â Â Â Â Â Â Â Â Â </span></span></strong><!--[endif]--><strong><span style="font-size: 10.0pt; line-height: 150%; font-family: 'Cambria','serif'; mso-ascii-theme-font: major-latin; mso-hansi-theme-font: major-latin;">Background: </span></strong><span style="font-size: 10.0pt; line-height: 150%; font-family: 'Cambria','serif'; mso-ascii-theme-font: major-latin; mso-hansi-theme-font: major-latin;">Difficulty in reaching an aetiological diagnosis and management issues are more in XY Disorders of Sex Development (DSD) than XX DSD. Clinical features are also similar between various aetiological types. </span></p> <p class="MsoListParagraphCxSpMiddle" style="margin: 0in; margin-bottom: .0001pt; mso-add-space: auto; text-align: justify; text-justify: inter-ideograph; text-indent: 0in; line-height: 150%; mso-list: l0 level2 lfo1;"><!-- [if !supportLists]--><strong><span style="font-size: 10.0pt; line-height: 150%; font-family: 'Cambria','serif'; mso-ascii-theme-font: major-latin; mso-fareast-font-family: Cambria; mso-fareast-theme-font: major-latin; mso-hansi-theme-font: major-latin; mso-bidi-font-family: Cambria; mso-bidi-theme-font: major-latin;">1.2.<span style="font-variant-numeric: normal; font-weight: normal; font-stretch: normal; font-size: 7pt; line-height: normal; font-family: 'Times New Roman';">Â Â Â Â Â Â Â Â Â Â Â Â </span></span></strong><!--[endif]--><strong><span style="font-size: 10.0pt; line-height: 150%; font-family: 'Cambria','serif'; mso-ascii-theme-font: major-latin; mso-hansi-theme-font: major-latin;">Objective</span></strong><span style="font-size: 10.0pt; line-height: 150%; font-family: 'Cambria','serif'; mso-ascii-theme-font: major-latin; mso-hansi-theme-font: major-latin;">: To find out the aetiology, clinical profile and management dilemmas of transgender children having Y containing chromosomes. </span></p> <p class="MsoListParagraphCxSpMiddle" style="margin: 0in; margin-bottom: .0001pt; mso-add-space: auto; text-align: justify; text-justify: inter-ideograph; text-indent: 0in; line-height: 150%; mso-list: l0 level2 lfo1;"><!-- [if !supportLists]--><strong><span style="font-size: 10.0pt; line-height: 150%; font-family: 'Cambria','serif'; mso-ascii-theme-font: major-latin; mso-fareast-font-family: Cambria; mso-fareast-theme-font: major-latin; mso-hansi-theme-font: major-latin; mso-bidi-font-family: Cambria; mso-bidi-theme-font: major-latin;">1.3.<span style="font-variant-numeric: normal; font-weight: normal; font-stretch: normal; font-size: 7pt; line-height: normal; font-family: 'Times New Roman';">Â Â Â Â Â Â Â Â Â Â Â Â </span></span></strong><!--[endif]--><strong><span style="font-size: 10.0pt; line-height: 150%; font-family: 'Cambria','serif'; mso-ascii-theme-font: major-latin; mso-hansi-theme-font: major-latin;">Materials and methods</span></strong><span style="font-size: 10.0pt; line-height: 150%; font-family: 'Cambria','serif'; mso-ascii-theme-font: major-latin; mso-hansi-theme-font: major-latin;">: A hospital based descriptive study was conducted in children with DSD from birth to 12 years of age who were attending pediatric and pediatric surgery outpatient and wards and endocrinology clinic of a tertiary care teaching hospital of south India for one year. Among them, those a Y containing chromosomes were analyzed for clinical features; investigated for arriving at a definitive diagnosis and management dilemmas were analyzed. Their gender identities were assessed. </span></p> <p class="MsoListParagraphCxSpMiddle" style="margin: 0in; margin-bottom: .0001pt; mso-add-space: auto; text-align: justify; text-justify: inter-ideograph; text-indent: 0in; line-height: 150%; mso-list: l0 level2 lfo1;"><!-- [if !supportLists]--><strong><span style="font-size: 10.0pt; line-height: 150%; font-family: 'Cambria','serif'; mso-ascii-theme-font: major-latin; mso-fareast-font-family: Cambria; mso-fareast-theme-font: major-latin; mso-hansi-theme-font: major-latin; mso-bidi-font-family: Cambria; mso-bidi-theme-font: major-latin;">1.4.<span style="font-variant-numeric: normal; font-weight: normal; font-stretch: normal; font-size: 7pt; line-height: normal; font-family: 'Times New Roman';">Â Â Â Â Â Â Â Â Â Â Â Â </span></span></strong><!--[endif]--><strong><span style="font-size: 10.0pt; line-height: 150%; font-family: 'Cambria','serif'; mso-ascii-theme-font: major-latin; mso-hansi-theme-font: major-latin;">Results: </span></strong><span style="font-size: 10.0pt; line-height: 150%; font-family: 'Cambria','serif'; mso-ascii-theme-font: major-latin; mso-hansi-theme-font: major-latin;">Causes could be identified in 82.3% cases. 53% of cases were Androgen Insensitivity Syndrome (AIS). 71.5% of children are being reared as female. 48% of them are under irregular follow-up. Major reason being social stigma. </span></p> <p>Â </p> <p class="MsoListParagraphCxSpLast" style="margin: 0in; margin-bottom: .0001pt; mso-add-space: auto; text-align: justify; text-justify: inter-ideograph; text-indent: 0in; line-height: 150%; mso-list: l0 level2 lfo1;"><!-- [if !supportLists]--><strong><span style="font-size: 10.0pt; line-height: 150%; font-family: 'Cambria','serif'; mso-ascii-theme-font: major-latin; mso-fareast-font-family: Cambria; mso-fareast-theme-font: major-latin; mso-hansi-theme-font: major-latin; mso-bidi-font-family: Cambria; mso-bidi-theme-font: major-latin;">1.5.<span style="font-variant-numeric: normal; font-weight: normal; font-stretch: normal; font-size: 7pt; line-height: normal; font-family: 'Times New Roman';">Â Â Â Â Â Â Â Â Â Â Â Â </span></span></strong><!--[endif]--><strong><span style="font-size: 10.0pt; line-height: 150%; font-family: 'Cambria','serif'; mso-ascii-theme-font: major-latin; mso-hansi-theme-font: major-latin;">Conclusion: </span></strong><span style="font-size: 10.0pt; line-height: 150%; font-family: 'Cambria','serif'; mso-ascii-theme-font: major-latin; mso-hansi-theme-font: major-latin;">Majority of cases of DSDs containing a Y containing chromosome is Androgen Insensitivity Syndrome. Majority of them being reared as females only. Those who are being reared as males will be either not having testis or dysgenetic testis or may have an abnormal looking male genitalia with or without hypospadias. Due to social stigma majority are under irregular follow up.</span></p>
Introduction
Intersex disorders are among some of the most distressing problems encountered at birth because of the uncertainty that clouds the newborn’s most basic identity. It is a social emergency requiring rapid determination of a definitive diagnosis to facilitate the development of a suitable treatment plan that will minimize medical, social and psychological complications [1]. Majority of XX DSD will be Congenital adrenal hyperplasia but the difficulty in reaching a diagnosis is more in XY DSD than XX DSD because of the variety of aetiological conditions and limitation of the diagnostic tests available. The management is more miserable due to the inability in constructing a male external genitala. So most often the disparity between genotypic and phenotypic sex is more in XY DSD. Humiliation created at the time of diagnosis will make the parents forget about the Pandora”s box in adolescence. Due to the social stigma attached, the first contact with the physician may also be the last. In this The research protocol of the study was submitted to the Research and Human ethical committee of the institution. The study was started after getting the clearance from both the committees. Children from birth up to 12 years of age, who fulfilled the criteria for DSD (Disorders of sex development) according to the standard literature Table 1 and who came to our paediatric outpatient (OP) clinic and wards, paediatric surgery OP and wards, and endocrinology clinic during one year period (1st January 2010 to 31st December 2010) were analyzed and those with a Y containing chromosome were enrolled. 21 cases were enrolled into the study.
context we tried to analyze the clinical profile of transgender children with a Y containing chromosomes and also tried to formulate a protocol to reach at a reasonable diagnosis in resource limited settings. We also peeped into the follow up issues as well.
Materials and Methods
| S. No | Disorders |
| 1 | A phallus and bilaterally non palpable testis |
| 2 | Unilateral cryptorchidism and hypospadias |
| 3 | Penoscrotal or perineoscrotal hypospadias with or without microphallus even if the testis are descended. |
| 4 | Discordance of external genitalia compared with prenatal karyotype. |
| 5 | Apparently female appearance with enlarged clitoris or inguinal hernia. |
| 6 | Overt genital ambiguity such as cloacal extrophy. |
Table 1: Disorders of Sex Development.
Table 1: Disorders of Sex Development. A proforma was prepared with personal data including age at first presentation and current age, assigned gender at birth, sex of rearing, gender identity, consanguinity, antenatal history and drugs, family history, initial presenting complaints, description of genitalia at presentation (from old records also), initial investigations - laboratory/ karyotyping/ imaging/ laparoscopy/ biopsy), initial diagnosis and treatment received (medical/ surgical), current appearance of external genitalia (prader staging), reinvestigations done , revised diagnosis , treatment planned, follow up pattern (regular/ irregular) and reason for irregular follow up [2]. The genital ambiguity inclusive of the symmetry of the labioscrotal folds, labial fusion, phallic length, morphology of the phallus, number of urogenital openings, palpable gonads, site of the gonads, position of urethral opening, chordee and anogenital ratio were recorded to describe the stage of virilisation. Laboratory investigations included serum electrolytes, random blood sugar, renal function, hormonal studies like basal concentration of 17– hydroxy progesterone (17-OHP), testosterone, Dihydrotestosterone (DHT), Dehydroepiandrosterone (DHEA), Follicle Stimulating Hormone (FSH) and Luteinising Hormone (LH) according to the clinical diagnosis. In cases with a significant testosterone level, hCG stimulation test was done after 3 months of age by giving 1000 units IM on alternate days for 3 doses to study the levels of testosterone and DHT after 3 months of life (this was postponed as a normal gonadotropic surge occurs with a resultant increase in the testosterone level and its precursors in early days). A failure to respond to hCG in combination with elevated LH and FSH levels was consistent with anorchia. A twofold or more increase in levels of testosterone2 or a level more than 300 ng/dl after 24 hours of last dose of hCG suggested the presence of functional testicular tissue [3]. A testosterone to DHT ratio along with androgen precursors before and after hCG stimulation helped to elucidate the 46, XY DSD etiology as shown in Figure 1 and Figure 2.
Karyotyping was done for all patients on peripheral blood lymphocytes. Pelvic and abdominal ultra sonography helped to detect the presence of gonads and mullerian structures. Other investigations included genitogram, laparoscopy and gonadal biopsy. For older cases, previous investigations were analysed, PCR for SRY or mutation analysis for enzyme defects were not available in our settings from the clinical examination and the investigations, and we arrived at the aetiological diagnosis of XY DSD as shown in Figure 1 and 2, which is made with modifications from standard literature. The definitions used for various diagnosis and abbreviations are given in Table 2.
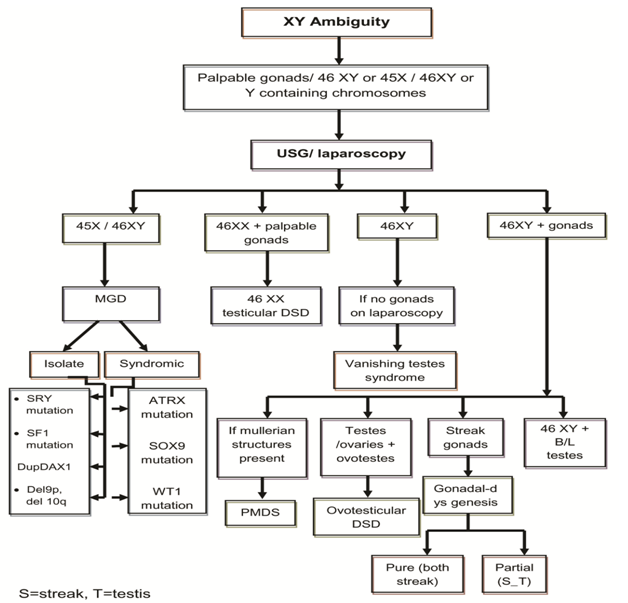
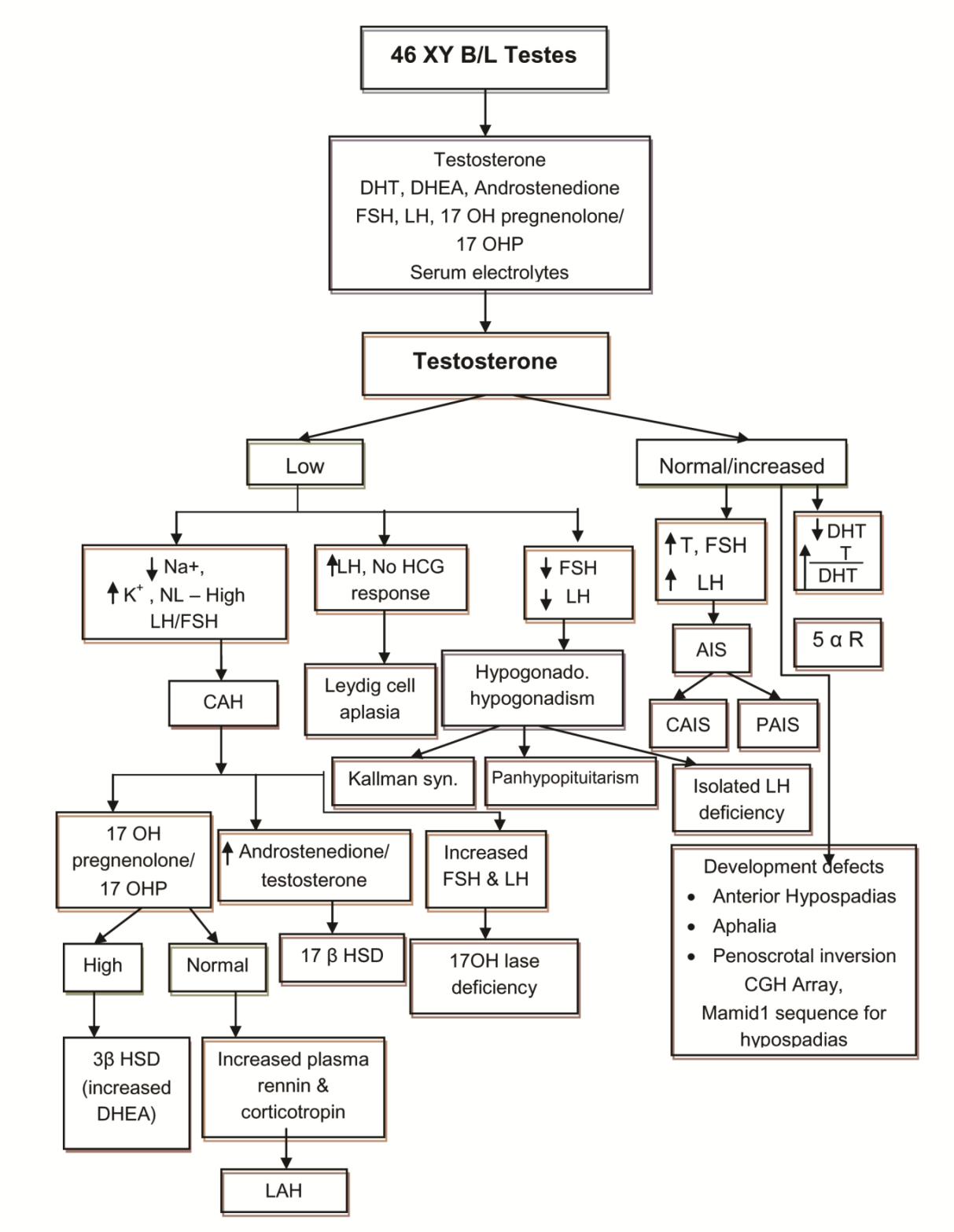
Diagnosis and Abbreviations Study
Ambiguous genitalia When there is difficulty in assigning sex to an individual from the appearance of external
- 46, XY DSD
- DSDs in which chromosome is 46 XY, testes may be normal or dysgenetic, the internal ducts system or the external genitalia are incompletely masculinized or female type
- 46, XY child with a female phenotype and testes are located internally or in inguinal canal.
- CAIS
- Normal/elevated basal testosterone, good hCG response of testosterone and absence of
- Varying degrees of undervirilisation of 46, XY individuals (penoscrotal/perineal hypospadias,
- Partial Androgen
- Insensitivity Syndrome undescended testis with hypospadias). Normal/elevated basal testosterone, good hCG response of testosterone and absence of mullerian structures on imaging but normal T/DHT (PAIS)
- These patients have a 46, XY karyotype and ambiguous external genitalia but normally
- 5 α reductase deficiency differentiated testes with male internal ducts. A testosterone to DHT ratio of greater
- 46, XY child with streak gonads and low levels of Testosterone. They have ambiguous development of the internal genital ducts, the urogenital sinus and the external genitalia.
- XY Pure gonadal dysgenesis (PGD)
- Affected 46, XY children have phenotypic variability from normal boys to normal girls.
- PMDS
- 46, XY with persistence of mullerian duct derivatives in otherwise completely virilized
- Ovotesticular DSD
- Children with expression of both ovarian and testicular tissue either in the same or in opposite gonads. The ovarian tissue is normal, but the testicular tissue is dysgenetic [10]. boys with classic XX with apparently normal male phenotypes, boys with non-classic XX
- 46, XX testicular DSD with some degree of sexual ambiguity, and XX ovotesticular DSD.
- Sex chromosome DSD: associated abnormality in sex chromosome
Table 2: Definitions Used In the Study.
- mullerian structures on imaging level. than 20 after hCG stimulation test [10]. males. Normal testosterone levels
- Age of First Presentation
- Among 21 cases 12 cases were diagnosed in < 1year and 9 cases later than 1 year. Late presentations were 8 cases of androgen sensitivity syndrome and one case of ovotesticular DSD.
- < 1 year 1year -5 year >5 year
- CAIS
- 3
- 3
- PAIS
- 1
- 1
- 1
- 5 α reductase deficiency
- 3
- PGD
- 1
- PMDS
- 1
- Ovotesticular DSD
- 1
- 1
- Abnormal karyotype
- 2
- Unidentified
- 3
Table 3: Age of first presentation.
Clinical Profile
Each one had more than one complaint at the time of presentation.
| Ambiguous Genitalia | Hypospadias | Cryptorchidism | Microphallus | Clitoro-omegaly | Inguinal swelling | Sibling History | |
|---|---|---|---|---|---|---|---|
| CAIS(6) | 6 | 2 | |||||
| PAIS(3) | 1 | 1 | 2 | 2 | 2 | ||
| 5 α R Deficiency(3) | 2 | 3 | 1 | 2 | |||
| PGD(1) | 1 | 1 | 1 | ||||
| PMDS(1) | 1 | 1 | |||||
| Workup incomplete(3) | 1 | 2 | 3 | ||||
| Ovotesticular DSD(2) | 1 | 1 | 2 | ||||
| Abnormal karyotype (2) | 1 | 1 | 2 | 1 |
Table 4: Clinical features.
Gender Outcome
| Chromosomal sex | Assigned sex | Phenotypic sex | |||
| (n=21) | male | female | transgender | Male(6)28.5% | Female(15)71.5% |
| CAIS(6) | 6 | 6(100%) | |||
| PAIS(3) | 2 | 1 | 3(100%) | ||
| 5αR Deficiency(3) | 1 | 2 | 3(100%) | ||
| PGD(1) | 1 | 1(100%) | |||
| PMDS(1) | 1 | 1(100%) | |||
| Workup incomplete(3) | 2 | 1 | 3(100%) | ||
| Ovotesticular | 46XX/46XY | 1 | 1(50%) | ||
| DSD | 46XY | 1 | 1(50%) | ||
| Abnormal karyotype | 47XXY | 1 | 2(100%) | ||
| Abnormal karyotype | 49XXXX(50)/48XXY(30)/46XY(20) | 1 |
Table 5: Gender outcome in DSDs with Y containing chromosomes.
| Social stigma | 80% |
| Unaware of need | 30% |
| Financial problems | 40% |
| Lack of family support | 40% |
| Lost confidence in treatment | 30% |
Table 6: Follow up.
| Maintaining vaginal anatomy | 62.5% (n=5) |
| Hypospadias | 37.5% (n=3) |
Table 7: Reasons for irregular follow up (n=10).
Discussion
In 2006, the Lawson Wilkins Pediatric Endocrine Society (LWPES) and the European Society for Pediatric Endocrinology (ESPE) published a consensus statement on management of intersex disorders and proposed the term ‘Disorders of Sex Development (DSD) .The new DSD classification includes three main diagnostic categories: sex chromosome DSD, 46, XY DSD (formerly male Pseudo hermaphrodite) and 46, XX DSD (formerly female Pseudo hermaphrodite) [11, 12, 13]. The category of sex chromosome DSD embraces not only ovotesticular DSD (formerly true hermaphroditism) and 45, X/46, XY mixed gonadal dysgenesis, but also Turner’s syndrome and Klinefelter’ssyndrome. Among 21 cases of DSDs containing Y chromosomes, 17 children were XY DSD, 4 were sex chromosome DSD.
In the literature, definite cause is identified only in 50% cases of XY, DSD. In our study causes were identified in 82.3% of cases with a reasonable accuracy. But some of the patients were not labeled a diagnosis before enrolling into the study. They were counseled several times to complete the investigations and to arrive at a definite diagnosis. The commonest cause of XY, DSD in our study was AIS (53% of cases (35.29% CAIS, 17.6% PAIS)) followed by 5- α reductase deficiency (17.67%). 5.9% (n=1) cases were there each in Pure gonadal dysgenesis (PGD) and persistent mullerian duct syndrome (PMDS). In the study by Joshi et al, also 28% of cases were Androgen insensitivity syndrome (19.2% PAIS, 8.7% CAIS), 22.8% were 5- α reductase deficiency, 3.5% had PGD, 1.8% had PMDS, and 15.8% cases had incomplete workup [14]. Other studies by Morel et al and Saage et al also showed 90% and 88% cases with AIS [15, 16]. Among AIS, CAIS constituted the majority (35.29%). We had 6 cases of CAIS. All were presented with complete female phenotype with inguinal swelling. 2 were evaluated and diagnosed early as their siblings presented with PAIS. There were 3 cases of Partial androgen insensitivity syndrome. 2 cases presented with clitoromegaly and inguinal swelling .One case with hypospadias and indeterminate sex. Karyotyping revealed 46, XY and investigations revealed elevated testosterone but normal T/DHT ratio even after hCG stimulation. Another 3 cases who presented with ambiguous genitalia /cryptorchidism and hypospadias were found to have low DHT values and high T/DHT ratio and diagnosed to have 5- α reductase deficiency. Differentiating PAIS from 5- α reductase deficiency is important as the latter group will masculinize at puberty presumably due to direct action of testosterone of the phallus [17]. One case of pure gonadal dysgenesis was a phenotypic female for whom there was ambiguity of genitals due to clitoromegaly and right inguinal swelling, who presented at the age of 3 months. Karyotype was 46, XY and ultrasound and laparoscopy revealed rudimentary uterus with bilateral fallopian tubes. Testicular biopsy revealed bilateral hyalinised testes. Gonadectomy and feminizing genitoplasty were done and now the child is being reared as female. There was one case of Persistent mullerian duct syndrome who was a phenotypic boy who had bilateral cryptorchidism from birth. But lost to follow up and later came back at 51/2 years of age with testis at the root of scrotum. Ultrasound revealed mullerian structures and inguinal area exploration revealed mullerian structures. The same was removed. The gonads were testes. The child is being reared as male only. Work up was incomplete in 3 cases (17.7%).One case was a newborn who presented with cryptorchidism and ambiguous genitals. Karyotyping was 46, XY. But the parents did not turned up for review later on. The other 2 cases were having cryptorchidism and hypospadias at birth, for which multiple surgeries were done. Investigations done could not point towards a definite aetiology. They were12 years of age on review and started undergoing testosterone therapy to increase the phallic size. Sex Chromosome DSD are rare in most of the studies. The Ovotesticular DSD is the rarest variant making only up to 10% of intersex disorders. About 70% have a 46,XX karyotype, fewer than 10% have 46, XY and about 20% have 46,XX/46,XY mosaicism the karyotypes were 46 XY and 46 XX/46 XY [18]. The 46 XY ovotesticular DSD baby presented with ambiguous genitalia at birth. On examination, the gonads were at the root of scrotum and biopsy revealed bilateral ovotestes. For the second case, the karyotype was 46 XX/46 XY. This is called Chimerism. 46 XX/46 XY chimer is an individual in whom some cells have the male chromosome complement and some have female. When more than one set of cell line with different sets of chromosomes make up the body, it is called Chimerism. It occurs when two non identical zygotes combine at a very early stage of development to form a single foetus. It is different from Mosaicism where the individual originates from single foetus [19]. In our study this case was a phenotypic girl who presented at the age of 4 years with left inguinal swelling. Gonadal biopsy revealed ovotestis with fibro muscular changes on the right side and testes on the left side. The abnormal karyotype of two patients was 47 XXY and 49 XXXXY (50) / 48 XXXY (30) / 46 XY (20). 47 XXY (Klinefelter Syndrome) presented with bilateral cryptorchidism, microphallus, developmental delay and abnormal facies in infancy. 49 XXXXY (50) / 48 XXXY (30) / 46 XY (20) is a rare karyotype and is considered to be a mosaic variant of klinefelter syndrome. In the literature we have not found any case reports of such a mosaicism. Our baby presented with ambiguous genitalia at birth. Had bifid scrotum with cryptorchidism and microphallus. He was a low birth weight baby with facial dysmorphism, cleft palate, clinodactyly, developmental delay, failure to thrive and congenital heart disease. Mosaic forms are due to mitotic non dysjunction after fertilization of the zygote. These forms can arise from a 46XY zygote or a 47 XXY zygote. Severity of somatic malformations is proportional to the number of additional X chromosomes. The classical triad of 49XXXXY syndrome consists of mental retardation, radio ulnar synostosis and hypogonadism. Other features included LBW, slow growth, cranial anomalies, abnormal genitals, multiple skeletal deformities with joint laxity and cardiac defects. When analyzing the presenting features, All of them had either inguinal swelling or cryptorchidism except 2 cases of 5 alfa reductase deficiency.
Gender Outcome
- In our study those XY children raised as females included CAIS, PAIS and pure gonadal dysgenesis. In a study by Gollu et al, those XY raised as females included CAIS, PAIS and 5 α reductase deficiency.
- All cases of CAIS were assigned female sex at birth and now also rearing as females. They have undergone vaginoplasty and testes were removed.
- Among PAIS, 66.6% were assigned female sex and
33.3% had an undetermined sex at birth but all are being reared as females. All of them underwent genital reconstruction procedures. 4. About 66.6% of 5 α reductase deficiency were assigned undetermined sex at birth and 33.3% were assigned male sex and all are being reared as males. 5. The case of PMDS was assigned male sex at birth and also raised as male. 6. XY pure gonadal dysgenesis was assigned female from birth. After gonadectomy and feminizing genitoplasty the child is being reared as female. 7. The work up incomplete cases of XY (n=3) were being raised as males. 8. Combining all only 28.5% of DSDs with Y containing chromosomes patients are being reared as males. So before assigning a sex to any XY DSD we should know the natural course of the aetiological type and its pubertal change and should try to reach a correct diagnosis as early as possible and till then no sex should be assigned. A rough guideline is as follows [18, 20, 21]. 9. All cases of CAIS should be reared as females as their external genitals are female looking and have female phenotype. But they won’t be a fertile female. Can have a sexual life after proper vaginoplasty. 10. In PAIS or gonadal dysgenesis-if phallic length is more than1.9cm and administration of testosterone enanthate/cypionate 25mg I/M 3-4 injections at 3-4 weeks intervals increases penile length to 2.5cm, rear as male. In a term infant if phallic size is less than 1.5 cm long and 0.7 cm wide, it is preferred to rear as female [22]. 11. Those XY DSD who are being reared as females should be provided with pre pubertal and pubertal estrogen therapy for a better looking female phenotype. They may need repeated genitoplasty and vaginoplasty and should be counseled regarding fertility issues. 12. Testicular biosynthetic defects like 17 ketosteroid reductase deficiency and 5αreductase deficiency, even if reared as female, will adopt a male gender role at puberty due to coincident increase in Testosterone and DHT to typical male levels & presumably due to direct action of testosterone of the phallus [17, 23, 24]. So they should be reared as male only [18]. 13. In Sex chromosome DSD, the 2 abnormal karyotypes are being reared as males. 49 XXXXY (50) / 48 XXXY (30) / 46 XY (20) is considered to be a mosaic variant of klinefelter syndrome. It is very rare to find such a mosaicism. Mosaic forms are due to mitotic non dysjunction after fertilization of the zygote. These forms can arise from a 46XY zygote or a 47 XXY zygote. 14. The 46 XY ovotesticular is being reared as male though the genitals are still ambiguous. The 46 XX/46 XYchimerism had undergone genital reconstruction and is being reared as female. 15. In Ovotesticular DSD, if highly virilised rear as male if there is good testicular function and no uterus. Otherwise remove the inappropriate gonad and rear as female. The main reason for irregular follow up is social stigma. Repeated surgeries were needed in 38%. Majority were for maintaining vaginal anatomy either for clitoromegaly or maintaining vaginal orifice. So in a nutshell, the treatment of DSD is incomplete and it become a chronic emergency. As most of them are from low socio economic and educational background, the first contact between the patient and the physician may also be the last.
Conclusion
It’s difficult to reach at a definite diagnosis in XY DSD as it is time consuming, unavailability of all investigations at ease, cost, need of perseverance from the part of parents. More difficult to follow them up due to different reasons. Thorough and apathetic counseling is essential from the first visit with detailed discussion with parents based on karyotype, endocrine function, genital anatomy, expected pubertal change and fertility issues to assign a sex for rearing. Androgen insensitivity syndrome is the most common XY DSD in our study. Majority of the DSDs containing Y chromosomes are being reared as female.
References
-
Committee on Genetics (2000) Evaluation of the newborn with developmental anomalies of the external genitalia. Pediatrics 106(1 pt 1): 138-142.
-
Allen L (2009) Disorders of sexual development. Obstet Gynecol Clin N Am 36(1): 25-45.
-
Garry LW (2008) Evaluation of a child with ambiguous genitalia: a practical guide to diagnosis and management. _In_: pediatric endocrine disorders. Meena PD, Vijayalaxmi B, Menon PSN (Eds) Orient Longman Ltd, pp: 401- 422.
-
Coran AG, Polley TZ (1991) Surgical management of ambiguous genitalia in the infant and child. J Pediatr Surg 26(7): 812-820.
-
Houk CP, Lee PA (2005) Intersexed states: Diagnosis and Management. Endocrinol Metab Clin North Am 34(3): 791-810.
-
Feldman KW, Smith DW (1975) Fetal phallic growth and penile standards for newborn male infants. J Pediatr 86(3): 395-398.
-
Parisi MA, Ramsdell LA, Burns MW, Carr MC, Grady RE (2007) A gender assessment team: experience with 250 patients over a period of 25 years. Genet Med 9(6): 348-357.
-
Bergada I, Milani C, Bedecarras P, Andreoneet L, Ropelato MG, et al. (2006) Time course of the serum gonadotropin surge, inhibins, and anti-mullerian hormone in normal newborn males during the first month of life. J Clin Endocrinol Metab 91(10): 4092- 4098.
-
Maclellan Dl, Diamond DA (2006) Recent advances in external genitalia. Pediatr Clin N Am 53(3): 449- 464 .
-
Hughes IA, Houk C, Ahmed SF, Lee PA (2006) Lwpes consensus group; espe consensus group. Consensus statement on management of intersex disorders. Arch Dis Child 91: 554-563.
-
Lambert SM, Vilain EJ, Kolon TF (2010) A practical approach to ambiguous genitalia in the newborn period. Urol Clin N Am 37(2): 195-205
-
Houk CP, Hughes IA, Ahmed SF, Lee PA (2006) Writing committee for the international intersex consensus conference participants. Summary of consensus statement on intersex disorders and their management. International intersex consensus conference. Pediatrics 118: 753-757.
-
Houk CP, Hughes IA, Ahmed SF, Lee PA (2006) International consensus conference on intersex organized by the lawson wilkins pediatric endocrine society and the european society for paediatric endocrinology. Consensus statement on management of intersex disorders. International consensus conference on intersex. Pediatrics 118: e488- e500.
-
Rajesh RJ, Sudha R, Meena PD (2006) Etiology and clinical profile of ambiguous genitalia an overview of 10 years experience. Indian Pediatr 43(11): 974- 978.
-
Morel Y, Rey R, Teinturier C, Nicolino M, Michel Cl, et al. (2002) Aetiological diagnosis of male sex ambiguity: a collaborative study. Eur J Pediatr 161(1): 49-59.
-
Savage MO, Chaussain JL, Evain D, Roger M, Caulorbe P, et al. (1978) Endocrine studies in male pseudohermaphroditism in childhood and adolescence. Clin Endocrinol 8(3): 219-231.
-
Patel ZM, Ambani LM, Dalal JJ, Gokarn VV, Desai MP, et al. (1983) Ambiguous genitalia. Indian Pediatr 20: 279-284.
-
Rajendran R, Hariharan S (1995) Profile of intersex children in south India. Indian Pediatr 32(6): 666- 671.
-
Yordam N, Alikasifoglu A, Kandemir N, Caglar M, Balci S, et al. (2001) True hermaphroditism: clinical features, genetic variants and gonadal histology. J Pediatr Endocrinol Metab 14(4): 421- 427.
-
Mouriquand PD (2004) Possible determinants of sexual identity: how to make the least bad choice in children with ambiguous genitalia. Bju Int 93 (3): 1- 2.
-
Berenbaum SA, Bailey JM (2003) Effects on gender identity of prenatal androgens and genital appearance: evidence from girls with congenital adrenal hyperplasia. J Clin Endocrinol Metab 88(3): 1102-1106.
-
Clayton PE, Miller WL, Oberfield SE, Ritzén EM, Sippell WG, et al. (2002) Consensus statement on 21-hydroxylase deficiency from the european society for paediatric endocrinology and the lawson wilkins pediatric endocrine society. Horm Res 58(4): 188-195.
-
Gollu G, Yildiz RV, Bingol Kologlu M, Yagmurlu A, Senyücel MF, et al. (2007) Ambiguous genitalia: an overview of 17 years experience. J Pediatr Surg 42(5): 840-844.
-
Thomas DF (2004) Gender assignment: background and current controversies. BJU Int 93(3): 47-50.
- Shaping Healthy Futures: Pediatric Endocrine Breakthroughs of 2025
- Precision Medicine in Obesity: Customizing Treatment for 2025
- The Thyroid Revolution: How 2025 is Redefining Hormone Health
- Editorial- Targeting Immunometabolism for Generating Innovative Therapies for Cancer
- Current Knowledge of Chickenpox
- Correlation of Preinjection Values of Gonadotropins and Estradiol Level with Clinical and Radiologic Evidence of Sufficient Pubertal Suppression in Girls with Central Precocious Puberty